Collection: Flexineb®
20 products
-
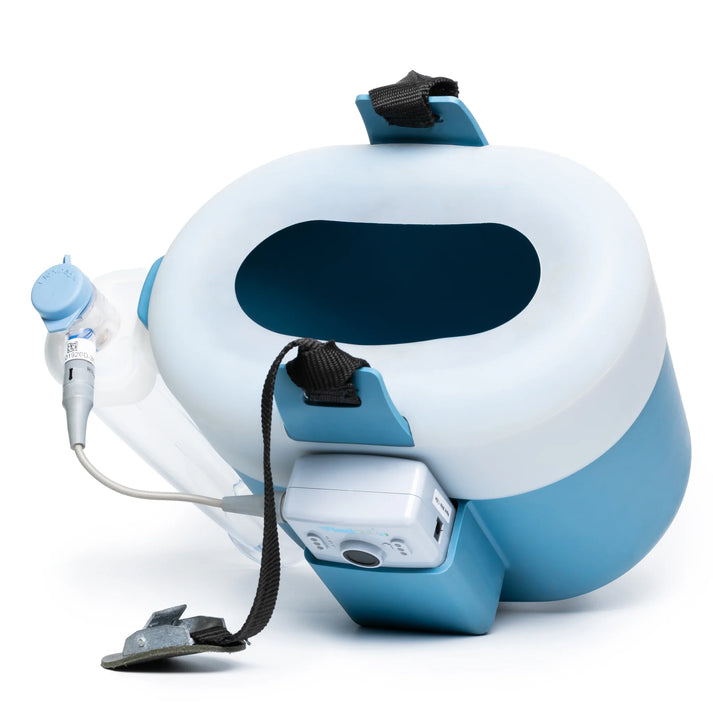
FlexiNeb® E3 - Includes 2 Med Cups and 8 oz SilvaPlex Natural Immune Support worth $19.50, Foal, Adult and Large Adult Sizes

 FlexiNeb® E3 - Includes 2 Med Cups and 8 oz SilvaPlex Natural Immune Support worth $19.50, Foal, Adult and Large Adult Sizes
FlexiNeb® E3 - Includes 2 Med Cups and 8 oz SilvaPlex Natural Immune Support worth $19.50, Foal, Adult and Large Adult Sizes- Regular price
-
$1,175.00 - Regular price
-
- Sale price
-
$1,175.00
Quick view
-
Mask Assembly with Aerosol Holding Chamber Kit (includes inlet valve), Exit Valve and Strap
 Mask Assembly with Aerosol Holding Chamber Kit (includes inlet valve), Exit Valve and Strap
Mask Assembly with Aerosol Holding Chamber Kit (includes inlet valve), Exit Valve and Strap- Regular price
-
$465.00 - Regular price
-
- Sale price
-
$465.00
Quick view
-
Soft Shell Mask with Exit Valve and Straps

 Soft Shell Mask with Exit Valve and Straps
Soft Shell Mask with Exit Valve and Straps- Regular price
-
$395.00 $435.00 - Regular price
-
- Sale price
-
$395.00 $435.00
Quick view
-
FlexiNeb® E3 Controller E3
 Vendor:FlexiNeb® E3 Controller E3FlexinebUS
Vendor:FlexiNeb® E3 Controller E3FlexinebUS- Regular price
-
$285.00 - Regular price
-
- Sale price
-
$285.00
Quick view
-
Box of 3 Extra Medication Cups

 Vendor:Box of 3 Extra Medication CupsFlexinebUS
Vendor:Box of 3 Extra Medication CupsFlexinebUS- Regular price
-
$210.00 - Regular price
-
- Sale price
-
$210.00
Quick view
-
FlexiNeb® E3 Soft Shell Mask
 FlexiNeb® E3 Soft Shell Mask
FlexiNeb® E3 Soft Shell Mask- Regular price
-
$190.00 $240.00 - Regular price
-
- Sale price
-
$190.00 $240.00
Quick view
-
Single Medication Cup

 Single Medication Cup
Single Medication Cup- Regular price
-
$80.00 - Regular price
-
- Sale price
-
$80.00
Quick view